
- Home
- Resarch
- Research Groups
- Prof. Feliu Maseras
Prof. Feliu Maseras
Improving the efficiency of catalytic processes by computational methods.


Research Overview
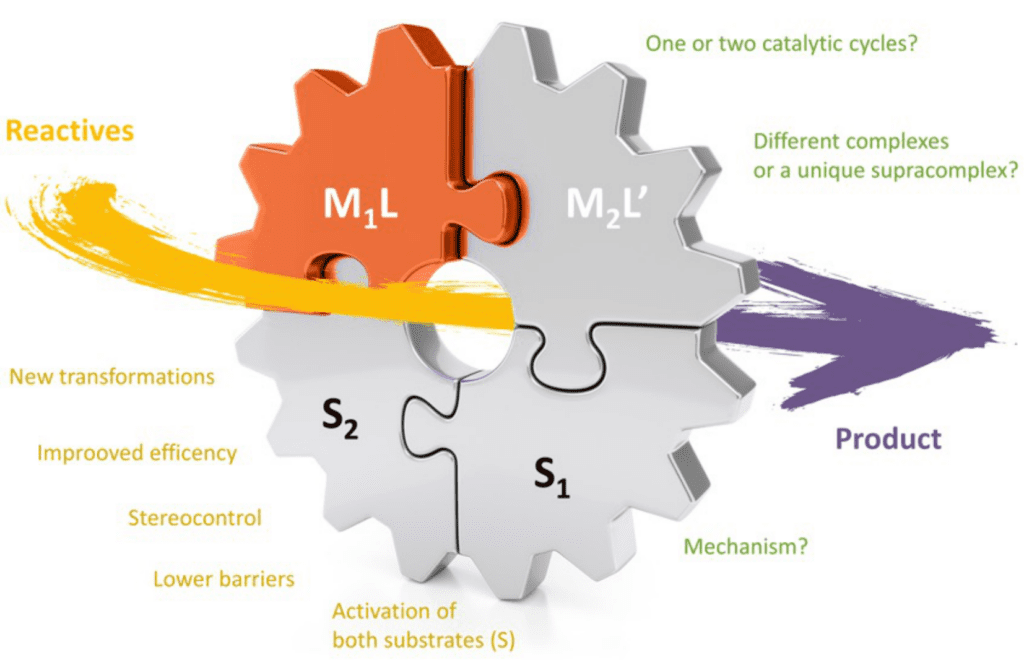
Computational homogeneous catalysis
Computational homogeneous catalysis has been traditionally the main focus of research in a group. In recent years, we have become interested in cooperative multimetallic catalysis. The simultaneous activation of two substrates or functionalities by different catalytic entities and their subsequent coupling are the responsible for the rate acceleration and selectivity exerted by these systems. The nature of the metal moieties is the responsible of the activation step, while selectivity mainly derives from supramolecular interactions between the substrates and the catalysts, particularly during coupling. The control of both activation and coupling opens the way for the design of new highly selective synthetic routes proceeding in mild conditions. Theoretical models provide fundamental insights on the substrate activation by metals as well as on the interactions between the different catalysts. This can allow the deciphering at the atomic level of the preferential pathways leading to different reaction products.
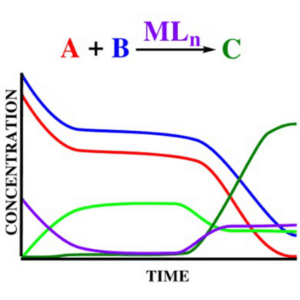
Concentration effects through microkinetic modeling
DFT models have been repeatedly demonstrated to be able to supply fundamental information on chemical processes through molecular insights into their mechanism and chemo-selectivity. However, the raw application of DFT free energy profiles falls shorts of reproducing the evolution of concentration of chemical species along the time, which is probably the most desirable quantitative information to compare calculation with the experimental data. In this context, microkinetic modelling emerges as the bridge between computed free energies and experimental data, allowing to obtain a theoretical kinetic profile of the chemical process directly comparable with experimental data. Our group in recent years has contributed to demonstrate that microkinetic modelling represents an essential tool in DFT-based mechanistic studies, from conventional organic and organometallic homogeneous catalysis to ball-milling mechanochemical reactions.
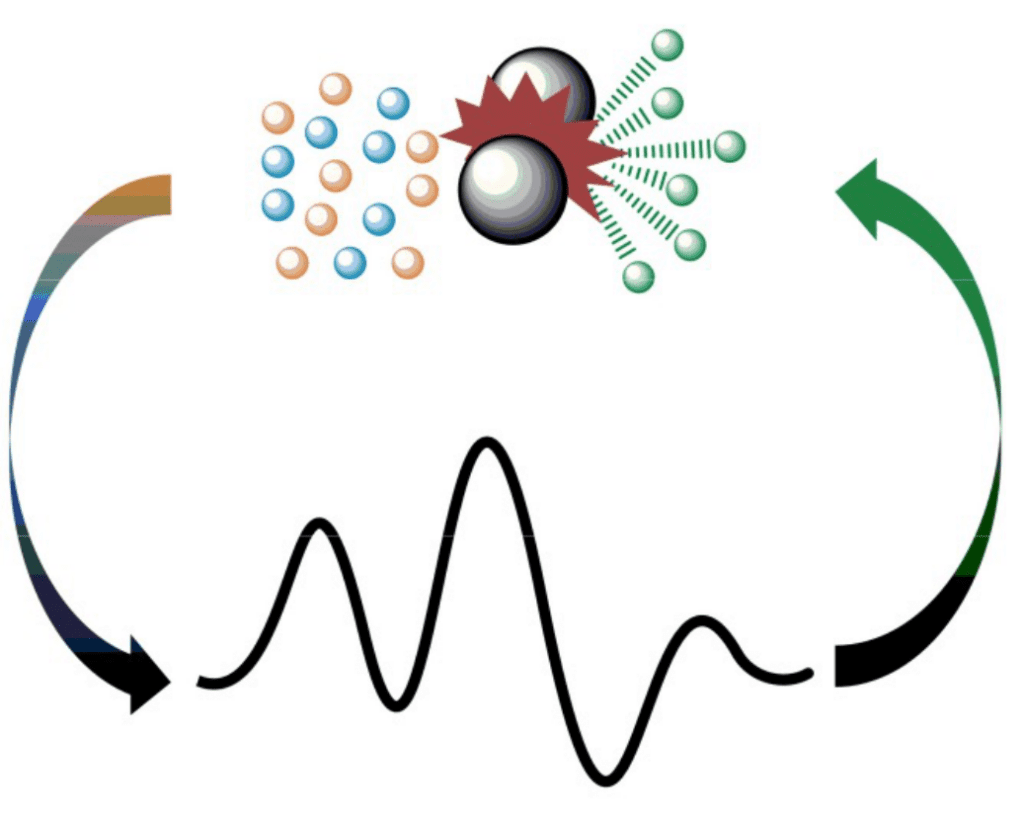
Computational mechanochemistry
The application of the computational chemistry techniques to the understanding and prediction of mechanochemical processes is gaining importance, even if still in the early stages. A complication for the systematization of these approaches is the variety of processes that are included under the label mechanochemistry. For this purpose, we classify the mechanochemical processes in two large groups: directional, where molecules are submitted to a single directional force; and isotropic, where the forces act in all directions on the molecules. Our initial results on the application of DFT and microkinetic modeling to ball-milling processes have been promising.
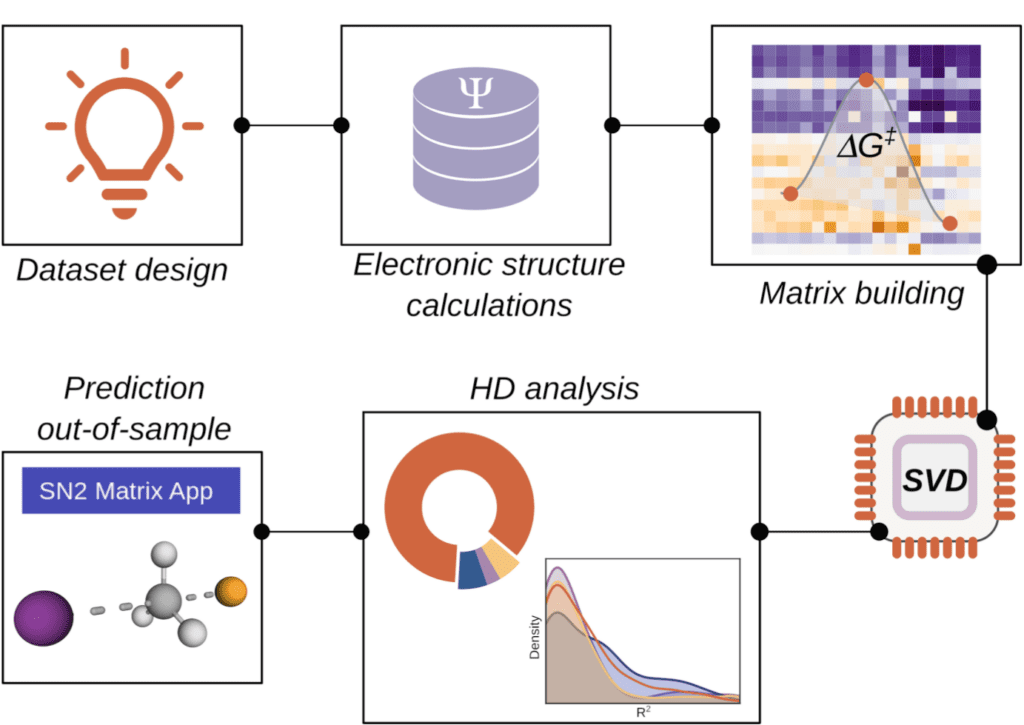
Hidden descriptors
The application of artificial intelligence to chemistry usually focuses on the identification of good correlations between descriptors and a given property of interest. The descriptors often come from arbitrary sets, with the implicit assumption that the evaluation of a sufficiently wide range of descriptors will lead to a satisfactory choice. Recent work in our group has focused on applying statistical analysis to large amounts of DFT results with the goal of finding optimal descriptor sets for a given property, which we label as hidden descriptors. With this treatment we have been able so far to gain chemical knowledge in two different domains: metal-ligand bond strength in transition metal complexes, and energy barriers in bimolecular nucleophilic substitution reactions. Ongoing research is being carried out on other chemical processes.

Let's create a brighter future
Join our team to work with renowned researchers, tackle groundbreaking
projects and contribute to meaningful scientific advancements

