
- Home
- Resarch
- Research Groups
- Prof. Carles Bo
Prof. Carles Bo
Computational modeling of chemical reactivity



Research Overview
Homogeneous Catalysis
Borylation & Diborations, metal catalysed and metal-free
In the 2009-2013 period, and in the framework of a long-term collaboration with Dr. Elena Fernandez from the URV, we continued exploring the catalytic activity of different metal complexes, borane reagents and unsaturated substrates, both experimentally and computationally. This collaboration consists in shared Ph.D. students that carry out half of their studies in the experimental laboratory (URV) and half in my computational laboratory (ICIQ).
Diboration of cyclic alkenes is not an easy transformation but palladium and platinum complexes achieve it with high selectivity. We demonstrated that N-heterocyclic carbene-pyridyl mixed ligands play a key role in activating the Pd(II) and Pt(II) complexes and thus cause a selective catalytic diboration reaction of the hindered cyclic alkenes, such as cyclooctene, cyclohexene, cyclopentene, norbornene and indene. [Link] One the most interesting advantages of this reaction when palladium is used is the possibility of a subsequent cross-coupling reaction. [Link]
Contrarily to rhodium-carbene complexes, which are also capable of diborating cyclic alkenes although less selectively, [Link] the reaction mechanism that we proposed involved a trans-metallation-like step instead of the classical oxidative addition that occurs for Rh(I). Indeed this fact suggested us for the first time that B-B diboration agents could be activated by the sole use of a base, and that a metal complex might not be needed.
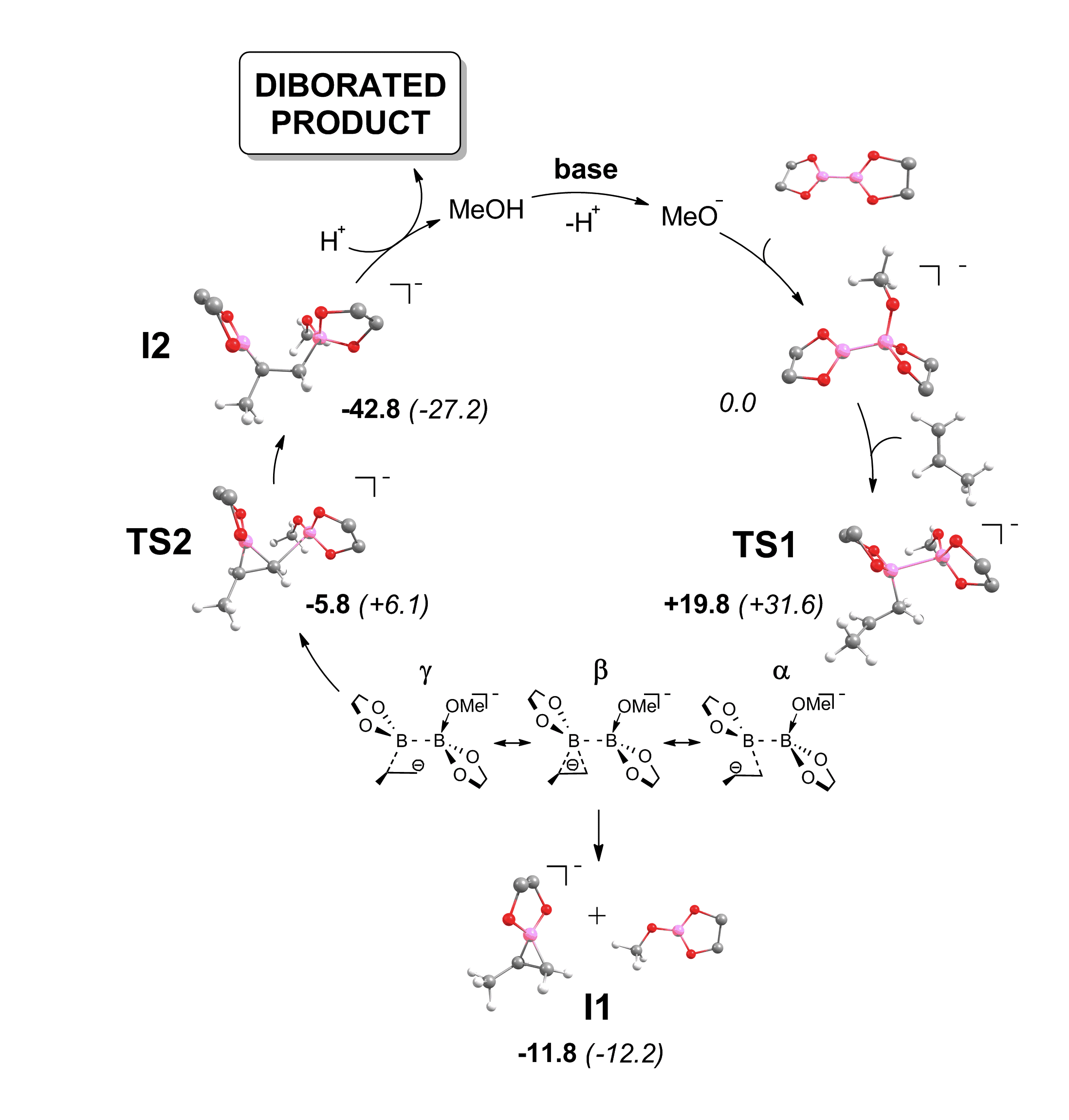
Figure 1. Proposed catalytic cycle for the metal-free diboration of alkenes.
Pursuing these ideas derived from computational studies, we succeeded in performing the reaction between non-activated olefines with diborons, in the absence of transition metal complexes (Figure 1). Our studies confirmed that a boron nucleophile is formed upon interaction of tetraalkoxydiboranes and a Lewis base, and identified the interaction of the adduct with the alkene as an overlap between the strongly polarised B-B s-bond (HOMO) and the antibonding p* orbital (LUMO) of the C=C. Conceptually, the boron switches its usual electrophilic character to an unusual nucleophilicity, and forces the olefin to act as an electrophile. [Link] An interesting feature of this mechanism is that, although the nucleophilic boron atom attacks the C1 atom, it is bonded to C2 in the product. Importantly, when electronic effects stabilise the reaction intermediate (i.e. they increase their kinetic stability, and hence their lifetime) the “hydroboration” side reaction becomes competitive with the diboration reaction. Actually, this is a sort of bifurcation reaction sequence (R-TS1-TS2-I1,I2), which connects two transition states and two products.
The new understanding about the mechanism for non-activated alkene substrates under MeOH/base exclusive conditions brought us to discover that activated unsaturated substrates (ketones, esters, etc.) could be borated too.[Link] In the case of alfa/beta unsaturated substrates, the hydroborated minor product in Figure 2 is then the major product. The role of the base in the organocatalytic boron conjugate addition reaction was thus determined by experimental and theoretical methods. A plausible mechanism was proposed in which the role of the base is crucial to provide the activated diboron reagent. Last but not least, a first approximation to the mechanism of the alkenes diboration reaction with the phosphine/MeOH/base system allowed us to propose phosphonium zwitterionic intermediates, which were fully characterised, both experimentally and theoretically, by means phosphorous and boron NMR. [Link]
The basic principles of these new chemical reactions are now fully understood and our theoretical studies demonstrated common features in the diboration reactions of unactivated alkenes and the β-boration of activated olefins. We expanded the application of these principles to other substrates such vinyl epoxides and aziridines. [Link] The same principle for activating a Bpin moiety is now being applied to the generation of organoselenium compounds by using the selenium boron species PhSeBpin. We found that the simple approach of the α,β-unsaturated carbonyl substrate to the selenium boron species PhSeBpin, promotes the PhSe transfer from the reagent to the substrate, in a selective manner (Figure 2). Remarkably, this direct addition does not require the presence of any transition metal complex as catalyst or bases or even co-solvents such as MeOH, unlike other E-Bpin additions. [Link]
CO2 Fixation
A new project deals with the mechanisms of the transition metal catalysed reaction of CO2 with epoxides to produce cyclic carbonates and/or polycarbonates, in the framework of a collaborative project with Prof. Kleij’s group at ICIQ. We reported on the monometallic mechanism operating in the cyclic carbonates formation from propylene oxide and CO2 catalysed by a Zn(Salphen) complex [Link] and a detailed analysis of the more active [Al{amino-tris-(phenolate)}]/NBu4I binary system. [Link]
QSSR
In 2008, we presented a new QSAR method for correlating structure-activity aimed at predicting the enantiomeric excesses and absolute configurations of transition metal catalysed reactions by using our own developed DFT-based 3D descriptors. In this period, in collaboration with former group member Dr. J. Carbó (URV), we applied this method to correlate activities and e.e.’s in the asymmetric hydroformylation of styrene by Rh-diphosphane complexes [Link] with some success, although serious difficulties were found when we applied the method to copper-catalysed cyclopropanation of alkenes [Link]. Still room to explore the predictability of these methods to challenging processes.
Other punctual collaborations:
Otero, UCM. Hybrid scorpionate/Cp Ti, Zr and Nb complexes, [Link 1, Link 2]
Castillón, URV. Rhodium catalyzed hydroiminacylation of alkenes [Link]
van Leeuwen, ICIQ. Hydrogen transfer catalysts based on new rhodium-secondary phosphine oxides (SPOs) complexes. [Link]
Creaser, Loughborough University, UK. IM-MS spectrometry of Zn-salphen complexes. [Link]
Llobet, ICIQ. Redox properties of Ru complexes with varying number of NHC carbene ligands. [Link]
Melchiorre, ICIQ. Mechanism for organocatalytic asymmetric reactions. [Link]
Bonjoch, UB, Barcelona. Unified approach to the synthesis of alkaloids. [Link]
Polyoxometalates
In the 2009-2013 period, and in collaboration with my former group at URV, we have been reviewing the latest applications of computational methods to POMs chemistry (electronic structure, properties, reactivity, simulations), a field that we initiated almost twenty years ago. [Link 1, Link 2, Link 3, Link 4]
Two of the papers published in 2009 (Link 1, Link 2) were selected to illustrate covers. Both papers dealt with the famous ball-shaped nanocapsule [{[MoMo5O21(H2O)6]}12{Mo2O4(L)}30]N-, known as Mo132. In collaboration with Prof. Weinstock (BGU, Israel) we described that carboxylates depending on their size negotiate passage through the 3 nm diameter pores [Link]. With the same group, in 2012 we reported for the first time a chemical reaction occurring inside the nanocapsule: the hydrolysis of methyl-tert-buthyl-eter. Our computed reaction mechanism enabled proposing the existence of an unexpected alkene intermediate, which was indeed detected experimentally[Ref. 32]. Much activity is being devoted in my group to this nanocluster, the electronic structure of which we described at a DFT level for the first time [Link]. The other cover [Link] refers to the structure of encapsulated water clusters, which self-assemble in layers resembling platonic solids. Molecular dynamics simulations enabled reproducing the structure of those layers, and also studying their physico-chemical properties [Link 1, Link 2]. The same MD techniques were applied to smaller systems to analyse the distribution of cations [Link 1, Link 2] around POMs surface, and to dissect the nature of solvent effects [Link].

Figure 2. Cover pages from [Link 1] (right) and [Link 2] (left).
The most cited paper in this period (over 100 cites in 4 years) corresponds to a collaboration with Bonchio’s group (U. Padova, Italy) on the mechanism of water oxidation by a tetra-Ruthenium substitute polyoxometalate.[Link]

Figure 3. HOMO of U28 capsule. Cover page Ref 43
The group carried out the first theoretical studies on a new class of recently discovered Uranium derived POMs known as Uranyl-peroxides. These anionic molecular metal-oxides are formed from uranyl salts when treated with base and hydrogen peroxide, and were firstly reported by Burns (U. Notre Dame, USA) in 2005. They form nano-cages of varying size and shapes resembling fullerenes, and might be used to abate nuclear waste in aqueous media. We firstly discussed the effects of alkali cations as templates for the distinct observed topologies [Link], and reported the first DFT calculations on a whole nanocapsule, U20 [Link]. In collaboration with Nyman’s group (Oregon State U., USA), we succeeded in predicting the geometric structure of series of U28 nanocapsules with complex formula [M@M’4M’’12 (UO2(O2)1.5)28]15– (M=Ta, Nb; M’=K, Rb, Cs; M’’=Na, K). The computed electronic structure was in good agreement with electrochemistry experiments, and justified the nanocapsule destruction upon reduction [Link]. More recently, in collaboration with Nyman’s and Navrostky’s groups (USA), our theoretical studies fully justified thermochemical measurements on the stability of U28 encapsulating anionic species. This work produced a paper that reached a cover [Link].
Supramolecular Chemistry
Over the 2009-2013 period, my group acquired some experience in this field, in which we applied state-of-the-art DFT methods for determining structures, spectroscopic properties and reaction mechanisms. In collaboration with Mendoza’s group (ICIQ), we proposed the structure of a capsule based on the cyclotriveratrylene (CTV) scaffold functionalized with three tetrathiafulvalene derivatives (ex-TTF), which forms “ball and socket” complexes with C60 and C70 fullerenes, and we explained their UV-vis spectra [Link]. In collaboration with Ballester’s group (ICIQ), we predicted that [Rh(nbd)2]+ could be encapsulated inside a resorcin[4]arene-based host, which modifies the normal outcome of nbd hydrogenation [Link], and we explained this distinct reactivity by determining the reaction mechanism of that encapsulated species [Link]. Also, electronic CD spectra computed from our structures for Bisporphyrin tweezer receptors allowed assigning the chirality of those host-guest complexes [Link].
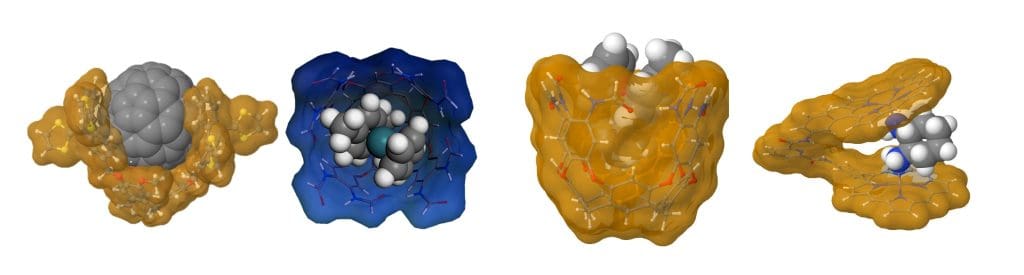
Figure 4. Some supramolecular host-guest complexes recently studied.

Let's create a brighter future
Join our team to work with renowned researchers, tackle groundbreaking
projects and contribute to meaningful scientific advancements

