
- Home
- Resarch
- Research Groups
- Prof. Antoni Llobet
Prof. Antoni Llobet
Molecular redox catalysis for the generation of solar fuels via artificial photosynthesis

Research Overview
Mother Nature uses a whole range of metalloproteins to carry out a wide variety of oxidation process in a highly selective and efficient manner. Thus, Llobet’s research group develops their transition metal redox catalysts inspired by nature’s structural motives and reactivity strategies.
Their catalyst development involves the interplay of a variety of disciplines including, synthetic organic and inorganic chemistry, spectroscopy, electrochemistry, kinetics and catalysis.
The following are some of the areas in which this group is involved:
Catalysts for Artificial Photosynthesis
The prediction for energy demand by the year 2050 is in the range of 30-50 TW which is more than a 100% increase with regard to what we have consumed by the year 2004. Further, given the rate of CO2 increase and the catastrophic consequences this is producing to our planet, it is obvious that our society has an urgent need for a carbon-neutral renewable energy source. An attractive and clean energy vector to solve this problem could be H2, but while the storage and separation of hydrogen has already been achieved with certain degree of success, the question of where do we get a sustainable hydrogen source still remains to be answered. Nature has been using water and sunlight as a source of energy in its photosynthetic processes for a long time. Oxidation of water to O2 is the terminal reaction of photosystem II (PSII) in green plants which takes place at a polynuclear Ca-Mn4 complex.
![]()
This reaction is a thermodynamically demanding reaction since Eº = 1.23 V (vs. SHE) at pH = 0.0. On the other hand it is of tremendous molecular complexity from a mechanistic perspective, since it involves the oxidation of water by 4 H+ and 4 e– with the formation of an oxygen-oxygen bond. It is thus an important reaction to be modelled since efficient low molecular weight models can lead to a first step towards creating a clean renewable energy source. The mastering of the performance of this reaction is nowadays the bottleneck for the development of artificial photosynthetic devices, capable of producing H2 from water and sunlight. Thus it constitutes the key to access a clean renewable fuel such as H2, the energy vector of the future.

Figure 1. Zero-emission fuel-cell bus.
Our group has recently prepared a family of dinuclear Ru-aqua complexes containing dinucleating polypyridylic ligands. These ligands are chosen because they enforce a particular metal preorganization and because they impose a delicate balance between the Ru-aqua entities. That is: they are sufficiently close so that they can interact to one another but at the same time they are sufficiently apart so that an oxo bridging reaction can not take place. The latter would be highly detrimental since it would basically kill the catalytic activity with regard to water oxidation.
The higher oxidation states of these Ru dinuclear complexes have been shown to be active with regard to the oxidation of water to molecular dioxyen. The group is currently studying the effects of electronic perturbations over the performance of the catalysts as well as the reaction mechanisms through which those reactions proceed. Further, they are designing specific ligands and complexes that will allow them to discriminate between potential intramolecular versus intermolecular mechanisms involving water nucleofilic attack to a Ruthenium-oxo electrofilic entity.
The high thermodynamic potential required for a catalyst capable of oxidizing water to molecular dixoygen implicates that its active species will also be capable of oxidizing a wide range of organic compounds. Therefore, intermolecular catalyst-catalyst deactivation is one of the main deactivation pathways. For this purpose the group is designing new dinucleating ligands that maintain the intrinsic electronic and coordination abilities of the original ligands but that contain a functionalized group that is able to be attached into an oxidatively rugged solid support. This will bring about a restricted translational mobility that would avoid those undesirable intermolecular interactions.
The combination of spectroscopic, electrochemical and mechanistic information allows them to design and develop new and more efficient water oxidation catalysts based on Ru complexes. Those complexes are to be integrated in complex solar cells in a serious first step towards a viable commercial application.
Our research group is also at the moment developing the parallel chemistry, substituting the Ru metal center by Mn, and modifying the ligands taking into account the specific chemistry of such a first raw transition metal. The aim here is to increase the velocity of the water oxidation process and to understand the reaction pathways through which they proceed.
Photochemical Oxidation of Organic Substrates
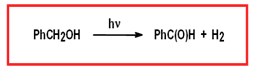
Our group is in the process of developing the adequate transition metal complexes that can be attached into TiO2 semiconductors, or alternatively into quantum dots, so that it can perform the oxidation of organic substrates with just sunlight. This reaction concept benefits from the absence of a generally polluting and expensive chemical oxidant and also from the formation of H2 as a by product.
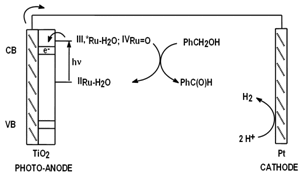
Figure 2. Proposed photo-oxidation scheme for organic substrates based on Ru complexes.
Selective Chemical Oxidation of Organic Substrates under Mild Conditions
There is an important number of dinuclear copper-containing proteins that use molecular dioxygen to carry out a variety of oxidations, including the industrially relevant oxidation of cyclohexane to cyclohexanol. In this topic the group is centering its efforts in the synthesis, characterization and reactivity of transition metal complexes using mononucleating and dinucleating ligands. The appropriate design of dinucleating ligands permits to situate two transition metals in relatively close proximity so that cooperation phenomena can take place as well as its fine tuning. These spatial effects combined with electronic effects, permit the generation of synergic phenomena and in some cases the viability of a given reaction that is not observable with a mononuclear complex. In nature a large number of metalloproteins exists, whose function consists in the breaking of O-O bonds under very smooth conditions,
![]()
for its subsequent incorporation in a specific substrate with high efficiencies and selectivity. As an example the oxidation of methane to methanol can be cited catalyzed by MMO (methane monooxigenase),
![]()
where AH2 represents an external reducing agent.
At present the chemical industry carries out a number of oxidation processes in a stoichiometric as well as in a catalytic manner but with low efficiencies and selectivity. Besides in general, expensive, highly toxic and environmentally harmful substances are used as oxidants. Here we deal with the field of the activation of oxygen utilizing models inspired in the molecular engineering of non-heme processes developed by nature, that is conceptually the inverse reaction of water oxidation.
The group’s objective is to elucidate the mechanisms through which these reactions occur in order to extract the key information to be able to design and build low molecular weight analogues. They are particularly interested in understanding the parameters that govern the intermolecular interaction between active species and external substrate. The key substrates they are dealing with at the moment are unsaturated hydrocarbons, alcohols, olefins and sulfides. With the last two ones they are also engaged in building up their corresponding chiral analogues to perform the catalytic oxidation in an enantioslective manner.
Supramolecular Catalysis
In this project molecular recognition is combined with oxidative catalysis in order to selectively functionalize a given substrate, a process that also occurs naturally. The challenge here is to prepare non-symmetric bifunctional molecules that are geometrically designed so that they can perform both tasks in a coordinated manner: the recognition process through a non covalent bond (coulombic interaction, hydrogen bonding, ?-stacking, etc.) and the oxidative process.
Activation of C-H and C-F Bonds
This project aims to understand the fundamental chemistry involved in activating C-H and C-F bonds by transition metal complexes. Selective activation of C-H bonds by metals under mild conditions is a major subject of research, which targets the final functionalization of organic substrates. It is essential to characterize the usually unstable intermediates of the C-H bond activation prior to functionalization if the mechanistics of the reaction are to be understood. This project, then, is of great importance in organometallic chemistry and catalyst development because it can deepen in our knowledge of the reactivity of stable bonds and the selective replacement of F atoms.

Let's create a brighter future
Join our team to work with renowned researchers, tackle groundbreaking
projects and contribute to meaningful scientific advancements

