
Straightforward computational determination of energy-transfer kinetics through the application of the Marcus theory
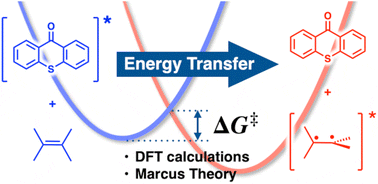
Energy transfer (EnT) photocatalysis holds the potential to revolutionize synthetic chemistry, unlocking the excited-state reactivity of non-chromophoric compounds via indirect sensitization. This strategy gives access to synthetic routes to valuable molecular scaffolds that are otherwise inaccessible through ground-state pathways. Despite the promising nature of this chemistry, it still represents a largely uncharted area for computational chemistry, hindering the development of structure-activity relationships and design rules to rationally exploit the potential of EnT photocatalysis. Here, we examined the application of the classical Marcus theory in combination with DFT calculations as a convenient strategy to estimate the kinetics of EnT processes, focusing on the indirect sensitization of alkenes recently reported by Gilmour, Kerzig and co-workers for subsequent isomerization [Z & auml;hringer et al., J. Am. Chem. Soc., 2023, 145, 21576]. Our results demonstrate a remarkable capability of this approach to estimate free-energy barriers for EnT processes with high accuracy, yielding precise qualitative assessments and quantitative predictions with typical discrepancies of less than 2 kcal mol-1 compared to experimental values and a small mean average error (MAE) of 1.2 kcal mol-1. Energy transfer barriers through affordable calculations. Density functional theory is shown to be a reliable tool to compute barriers between relatively large systems through the application of the classical formulation of the Marcus theory.
Sole-Daura, A.; Maseras, F.
Chem. Sci. 2024
DOI:
10.1039/d4sc03352c

- SHARE






Let's create a brighter future
Join our team to work with renowned researchers, tackle groundbreaking
projects and contribute to meaningful scientific advancements