
Electronic Properties of Single-Atom Metal Catalysts Supported on Nitrogen-Doped Carbon
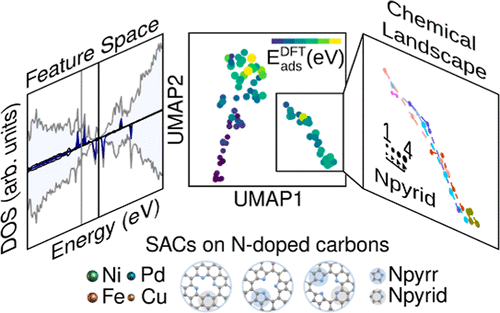
Single-atom serve as catalysts (SAC) with intriguing properties, offering a balance between heterogeneous and homogeneous properties. While their heterogeneous nature has been extensively explored, the role of the scaffold as a ligand remains obscure but severely affects the metal electronic structure. In this study, we employ density functional theory to investigate the electronic properties of single-atom catalysts supported on nitrogen-doped carbon, focusing on the extensively used transition metal centers: Fe, Ni, Cu, and Pd. Our analysis includes metrics such as Bader charges, the metal d-band center, and the Fermi level, focusing on the coordination environment of the transition metal center and analyzing the influence of the nitrogen content on these properties. We utilized dimensionality reduction techniques to identify similar electronic configurations. Our findings reveal that the SACs electronic structure predominantly exhibits metal cations with free-atom-like d-states, with the coordination environment’s significance outweighing the metal nature, and further influenced by nitrogen content and type. Ultimately, our study underscores the ability to finely tune the host to adapt to reactivity, offering insights into the continuous modulation of electronic structures.
Minotaki, M. G.; Lopez, N.
J. Phys. Chem. C 2024, 128, (33), 13816-13828
DOI:
10.1021/acs.jpcc.4c03232

- SHARE






Let's create a brighter future
Join our team to work with renowned researchers, tackle groundbreaking
projects and contribute to meaningful scientific advancements