
Decoding the CO2 Reduction Mechanism of a Highly Active Organometallic Manganese Electrocatalyst: Direct Observation of a Hydride Intermediate and Its Implications
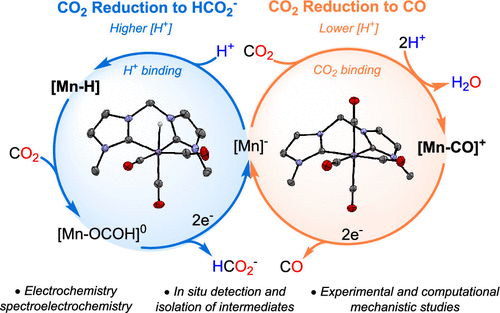
A detailed mechanistic study of the electrochemical CO2 reduction catalyzed by the fac-[MnI(CO)3(bis-MeNHC)MeCN]+ complex (1-MeCN+) is reported herein by combining in situ FTIR spectroelectrochemistry (SEC), synthesis and characterization of catalytic intermediates, and DFT calculations. Under low proton concentrations, 1-MeCN+ efficiently catalyzes CO2 electroreduction with long catalyst durability and selectivity toward CO (ca. 100%). The [Mn-I(CO)3(bis-MeNHC)]− anion (1–) and the tetracarbonyl [MnI(CO)4(bis-MeNHC)]+ complex (1-CO+) are key intermediates of the catalytic CO2-to-CO mechanism due to their impact on the selectivity and the reaction rate, respectively. Increasing the proton concentration increases formate production (up to 15% FE), although CO remains the major product. The origin of formate is ascribed to the competitive protonation of 1– to form a Mn(I) hydride (1-H), detected by SEC in the absence of CO2. 1-H was also synthesized and thoroughly characterized, including by X-ray diffraction analysis. Stoichiometric reactivity studies of 1-H with CO2 and labeled 13CO2 indicate a fast formation of the corresponding neutral Mn(I) formate species (1-OCOH) at room temperature. DFT modeling confirms the intrinsic capability of 1-H to undergo hydride transfer to CO2 due to the strong σ-donor properties of the bis-MeNHC moiety. However, the large potential required for the HCOO– release from 1-OCOH limits the overall catalytic CO2-to-HCOO– cycle. Moreover, the experimentally observed preferential selectivity for CO over formate is dictated by the shallow kinetic barrier for CO2 binding to 1– compared to the Mn–H bond formation. The detailed mechanistic study highlights the reduction potential, pKa, and hydricity of the metal hydride intermediate as crucial factors affecting the CO2RR selectivity in molecular systems.
Fernández, S.; Franco, F.; Martínez Belmonte, M.; Friães, S.; Royo, B.; Luis, J. M.; Lloret-Fillol, J.
ACS. Catal. 2023, 13, 10375-10385
DOI:
10.1021/acscatal.3c01430

- SHARE






Let's create a brighter future
Join our team to work with renowned researchers, tackle groundbreaking
projects and contribute to meaningful scientific advancements

