

 23/07/2019
23/07/2019
 12:00
12:00
- Lecturer: Prof. Dr. Wesley Browne
- University: University of Groningen (The Netherlands)
Moving Beyond Sensitised Photocatalysis - Light Driven Oxidation of with Molecular Based non-heme Iron Catalysts
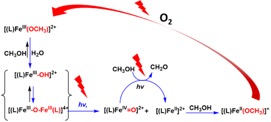
The photophysics and photochemistry are seeing a recent resurgence in interest due to the need to supplant photoactive complexes based on Ir(III) and Ru(II). However, the low lying 3MC states of iron complexes has in large part lead to the conclusion that iron complexes are not generally photoactive in themselves. Recently, however, we have shown that non-heme iron(II) complexes such as those based on pentadentate ligands, e.g., N4Py (N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine), undergo visible light driven oxidation to their iron(III) state in the presence of O2 without ligand degradation. Remarkably, however, the same complex in its higher oxidation states undergoes photochemical reduction with oxidation of solvent or organic substrates. This opposing photochemical reactivity allows for a complete cycle to be achieved in the catalytic activation of oxygen to oxidize methanol. Under mildly basic conditions, however, highly selective base catalysed ligand degradation with O2, to form a well-defined pyridyl-imine iron(II) complex and an iron(III) picolinate complex, is accelerated photochemically. Specifically a pyridyl-CH2– moiety is lost from the ligand yielding a potentially N4 coordinating ligand containing an imine motif. The involvement of reactive oxygen species other than O2 is excluded; instead deprotonation at the benzylic positions to generate an amine radical is proposed as the rate determining step. The selective nature of the transformation holds implications for efforts to increase catalyst robustness through ligand design. Taken together these data open up new opportunities in photocatalysis without the need for light harvesting photosensitisers.
-
Share
-

-

-

-

-

-

Other events

Let's create a brighter future
Join our team to work with renowned researchers, tackle groundbreaking
projects and contribute to meaningful scientific advancements