

 15/09/2017
15/09/2017
 12:00
12:00
 ICIQ Auditorium
ICIQ Auditorium
- Lecturer: Prof. Dr. Dennis G. H. Hetterscheid
- University: Universiteit Leiden (The Netherlands)
Iron Based Molecular Electrocatalysts for the Water Oxidation Reaction
Fundamental understanding regarding the water oxidation reaction is of key importance for our future chemical society. In my group the mechanism of electrochemical water oxidation mediated by molecular iron catalysts are studied by a plethora of in situ spectroscopy and characterization techniques coupled to electrochemistry to firmly establish which species are present at the electrode interface during the catalytic reaction. This allows us to convincingly connect the observed kinetics that were obtained under well-defined reaction conditions to the actual structure of the true active species, which is indispensable for drawing reliable conclusions concerning the catalytic mechanism. Moreover ambiguities by the sacrificial reagents that are typically used can are avoided in this approach.
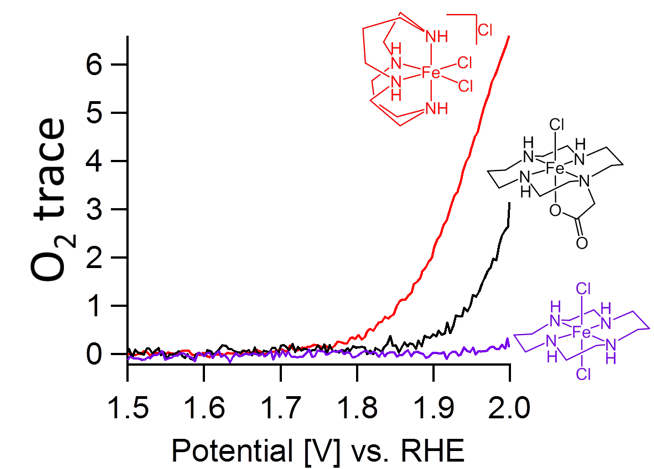
We have found that iron sites bearing cyclam ligands and cis-vacant sites are more potent water oxidation catalysts compared to their trans analogs with trans-vacant sites, in line with previous studies using sacrificial reagents (see Figure for on-line electrochemical mass spectrometry data). Besides a previous mentioned kinetic effect,1 wherein an iron bound hydroxide in that is capable of activating the water molecule for nucleophilic attack on an iron-oxo fragment in cis-position, subtle changes in the electronic structure of the catalysts were found to be of key importance. This caused that the iron catalyst with cis-vacant sites is substantially easier oxidized to the +V oxidation state that is necessary to split water.2
Based on these design principles, we now describe a new generation of catalysts that in contrast to the cyclam system is 1) more electron rich resulting in an oxygen evolution reaction at a much lower overpotential and 2) are considerably more stable under the required oxidative conditions since β-hydrogens, that can trigger ligand oxidation reactions, are rigorously avoided. Mechanistic studies regarding the precise reaction mechanism are in progress.
-
Share
-

-

-

-

-

-

Other events

Let's create a brighter future
Join our team to work with renowned researchers, tackle groundbreaking
projects and contribute to meaningful scientific advancements