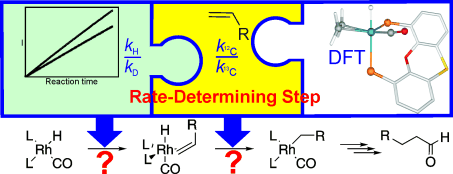
The rate-determining step in the hydroformylation of 1-octene, catalysed by the rhodium-Xantphos catalyst system, was determined by using a combination of experimentally determined 1H/2H and 12C/13C kinetic isotope effects and a theoretical approach. From the rates of hydroformylation and deuterioformylation, a small 1H/2H isotope effect of 1.2 was determined for the hydride moiety of the rhodium catalyst. 12C/13C isotope effects of 1.012(1) and 1.012(3) for the -carbon and -carbon atoms of 1-octene were determined, respectively. Both quantum mechanics/molecular mechanics (QM/MM) and full quantum mechanics calculations were carried out on the key catalytic steps, for real-world ligand systems, to clarify whether alkene coordination or hydride migration is the rate-determining step. Our calculations (21.4 kcal mol-1) quantitatively reproduce the experimental energy barrier for CO dissociation (20.1 kcal mol-1) starting at the (bisphosphane)RhH(CO)2 resting state. The barrier for hydride migration lies 3.8 kcal mol-1 higher than the barrier for CO dissociation (experimentally determined trend 3 kcal mol-1). The computed 1H/2H and 12C/13C kinetic isotope effects corroborate the results of the energy analysis.