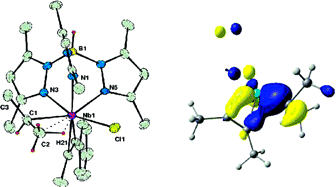
Density functional theory is used to explore the origins of the alpha- and beta-C-H agostic distortions in two isomers of TpMe2NbCl(MeCCPh)(i-Pr), both of which have been characterized experimentally. We first establish that our computational methodology provides a solid foundation on which to build such a model by showing that it reproduces a wide range of experimental observables, including structural, thermodynamic, and kinetic data and spin-spin coupling constants, 1JC-H, of the isopropyl ligand. Analysis of the converged electron density for the beta-agostic rotamer reveals that the molecular graph is highly dependent on functional: in some cases, bond and ring critical points can be located, while in others, they merge into a singularity, despite the fact that the associated changes in geometry are almost imperceptible. The unpredictable behavior of the molecular graph confirms that the electron density in the region between the metal and the C-H bond is almost homogeneous, but an accurate description of this region is clearly critical because only those functionals that conform to the uniform electron gas (UEG) limit are able to locate the beta-agostic minimum. Taken together, these observations suggest that through-space interactions between the C-H bond and the metal make an important contribution to the stability of the system. In contrast, the molecular graph for the alpha-agostic isomer is completely independent of functionals: no Nb···H critical point has ever been located. This, combined with the fact that the a-agostic structures optimized with UEG and non-UEG functionals are very similar, suggests that through-space interactions between the C-H s bond and the metal are less important in this case. Analysis of the Kohn-Sham and natural bond orbitals for the a-agostic isomer suggests that the distortion is instead driven by delocalization of the Nb-Ca sigma bonding electrons onto the unsaturated alkyne ligand. The underlying cause is the energy mismatch between the early-transition-metal d orbitals and the lone pair on the formally anionic alkyl ligand, which forces delocalization of the Nb-Ca bonding electrons onto the p* orbitals of the alkyne ligand. The same mechanism is also important in the beta-agostic system, although in this case, the agostic distortion is further stabilized by a through-space interaction between the beta-C-H s bond and the metal.