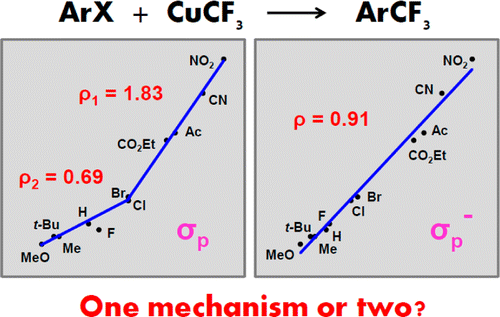
A combined experimental (radical clock, kinetic, Hammett) and computational (DFT, MM) study of the trifluoromethylation reaction of aryl halides with CuCF3 reveals a nonradical mechanism involving Ar–X oxidative addition to the Cu(I) center as the rate determining step. The reaction is second order, first order in each reactant with ΔG⧧ ≈ 24 kcal/mol for PhI (computed ΔG⧧ = 21.9 kcal/mol). An abrupt change in the gradient on the Hammett plot of log(kR/kH) versus σp for 11 p-RC6H4I substrates produces two correlations (ρ = +0.69 and +1.83), which is temptingly suggestive of two different reaction pathways. Only one mechanism is operational, however, as advocated by a single linear correlation with σp– (ρ = +0.91), analysis of the experimental ρ values, close similarity of the transition states varying in R and displaying clear signs of −M interactions, and excellent reproduction of the plot by DFT. The long-known yet previously uncomprehended ortho effect has been quantified, for the first time, using the reaction of CuCF3 with a series of o-RC6H4Br: R(kR/kH) = H (1) < Me (3.5) < MeO (4) < CN (20) < CHO (250) < CO2Me (850) < NO2 (4300) < Ac (7300) < CO2H (150 000). With minor contributions from electronic factors, the ortho effect is largely determined by (i) the stabilizing coordination of the o-substituent to Cu in the transition state with the Cu···O distance varying directly with the barrier and (ii) the steric bulk of the o-substituent that raises the ground state free energy of the haloarene (Goortho – GoH or Goortho – Gopara) by inflicting molecular strain and consequently weakening the Ar–X bond.