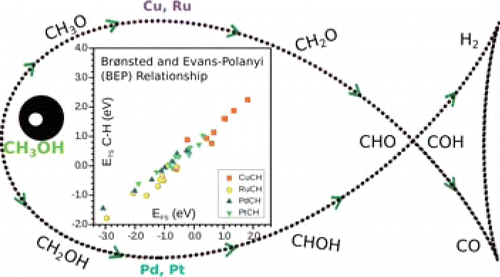
Methanol decomposition on metals has been subject of several theoretical studies, usually concentrating on a particular set of reactions in the main reaction path. In this work, we present an extensive study that considers all potential elementary steps for four close-packed surfaces including Cu, Ru, Pt, and Pd that shows the different behaviors and alternative routes through which the decomposition can take place by theoretical methods, including dispersion contributions. Decomposition follows different paths on these metals; while Cu would produce CH2O, CO is the major product for the other metals. In addition, coverage effects might change the first step in Pt and Ru from methylenic to alcohol H activation. Alternatively the reaction network can be inspected for the formation of methanol from CO and hydrogen. Under these conditions, Cu generates CH2O and only at very high H coverages is methanol likely to appear. On Pd, methanol formation and CHOH dissociation compete, thus leading to an inefficient process. A similar path takes place for Pt. For Ru the lateral paths leading to C–O breaking can occur at several points in the reaction network, never reaching CH3OH. A compilation of the results with comparable computational setups presents a detailed database that can be added to the thermodynamics and kinetics for other reactions, such as methanation, with which they share a common list of reactions, or employed when analyzing larger alcohols such as those derived from biomass.