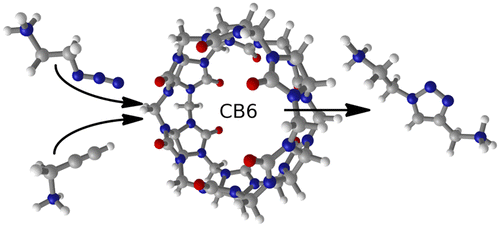
The enhanced reactivity of the Huisgen 1,3-dipolar cycloaddition between the protonated forms of azidoethylamine and propargylamine inside the cucurbit[6]uril host has been computationally studied. A DFT approach is applied to explore the relative stabilities and connections of a large variety of possible host–guest aggregates that may be formed in solution, as well as their reactivity. The free energies resulting from the DFT calculations are converted to rate and dissociation constants and introduced, together with the experimentally reported initial concentrations, in a kinetic simulation. The results reproduce the experimental observations and provide a detailed description of the behavior of a large host–guest system over time. The major cause of the rate acceleration inside the nanovessel is the reduction of the entropic component of the free energy barrier, and the existence of stable nonproductive host–guest adducts is identified as a major obstacle to improved catalysis.