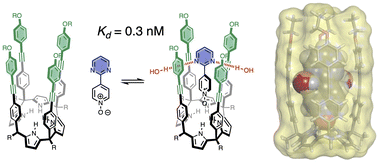
Conformationally well-defined supramolecular complexes that can be studied in different solvents provide a platform for separating and quantifying free energy contributions due to functional group interactions and desolvation. Here 1:1 complexes formed between four different calix[4]pyrrole receptors and eleven different pyridine N-oxide guests have been used to dissect the factors that govern aromatic interactions with heterocycles in water and in chloroform solution. 1H NMR spectroscopy shows that the three-dimensional structures of the complexes are fixed by four H-bonding interactions between the pyrrole donors at the bottom of the receptor and the N-oxide acceptor on the guest, locking the geometrical arrangement of interacting functional groups in the binding pocket at the other end of the receptor. An aromatic heterocycle on the guest makes two stacking interactions and two edge-to-face interactions with the side walls of the receptor. Chemical double mutant cycles were used to measure the free energy contribution of these four aromatic interactions to the overall stability of the complex. In chloroform, the aromatic interactions measured with pyridine, pyrimidine, furan, thiophene and thiazole are similar to the interactions with a phenyl group, but the effect of introducing a heteroatom depends on where it sits with respect to the aromatic side-walls of the cavity. A nitrogen lone pair directed into a pi-face of the side-walls of the binding site leads to repulsive interactions of up to 8 kJ mol-1. In water, the heterocycle aromatic interactions are all significantly more favourable (by up to 12 kJ mol-1). For the non-polar heterocycles, furan and thiophene, the increase in interaction energy correlates directly with hydrophobicity, as measured by the free energy of transfer of the heterocycle from n-hexadecane into water (Delta G degrees(water-hex)). For the heterocycles with polar nitrogen H-bond acceptors, water can access cracks in the walls of the receptor binding site to solvate the edges of the heterocycles without significantly affecting the geometry of the aromatic interactions, and these nitrogen-water H-bonds stabilise the complexes by about 15 kJ mol-1. The results highlight the complexity of the solvation processes that govern molecular recognition in water. Measurements using chemical double mutant cycles show that non-covalent interactions between heterocyclic and aromatic rings are more favourable in water than chloroform, provided polar sites are solvated by H-bonded water molecules in the complex.