Abstract
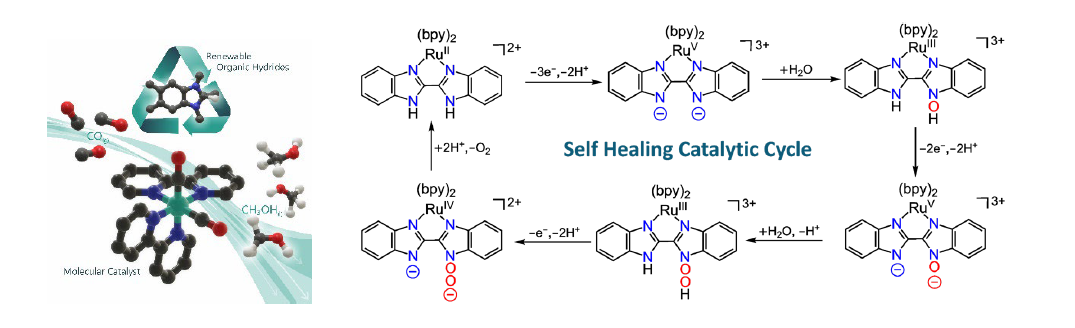
In nature, solar energy is captured and stored in chemical bonds by activating small molecules such as CO2 and H2O. This requires catalysts capable of carrying out CO2 reduction and water oxidation catalysis. In this presentation, molecular catalysts and mechanisms related to these two processes will be discussed. CO2 reduction to C1 products has received considerable attention as part of many efforts to try to address the devastating effects of climate change. Among various C1 reduction products, liquid fuels such as formic acid and methanol are particularly important to achieve carbon-neutral cycles. But the underlying chemistry to generate liquid fuels from CO2 efficiently and selectively has yet to be developed, particularly in the case of methanol. Cascade strategies using a combination of catalysts that can carry out some of the required multistep reactions are being pursued because achieving this goal with a single catalyst is difficult. One of these strategies involves CO2 reduction to CO by one catalyst and further reduction of CO to a liquid fuel by another catalyst or a combination of catalysts. In this presentation, a strategy for the generation of methanol efficiently and selectively from CO is discussed, with a transition metal-organic hydride combination. Using NMR, IR and mass spectrometry, together with labeling studies, chemical synthesis of authentic samples of intermediates and theoretical calculations, selective and efficient conversion of CO to methanol is demonstrated. The key to the high selectivity is the use of organic hydride donors that are inactive towards direct CO2 reduction and hydrogen generation. These organic hydride donors are inexpensive and renewable and are a promising platform for solar fuels generation from CO2. In the water oxidation front, a new mechanism for water oxidation catalysis with molecular catalysts will be presented. In this mechanism, the transition metal participates in the redox chemistry but not in the thermally activated processes, which take place on the ligands. In the oxygen evolution step, the ligands are returned to their original state and, as a result, these catalysts are very robust because they follow a “self-healing” mechanism. In addition, because the mechanism is ligand-based, no open coordination site is required and this enables the development of highly active molecular catalysts for water oxidation with first-row transition metals.